Most read this week
-
 Quantum dot laser set to transform medicine and communications
Quantum dot laser set to transform medicine and communications
-
 Turning industrial waste into clean hydrogen fuel
Turning industrial waste into clean hydrogen fuel
-
 Goldene, graphene’s golden cousin produced for the first time
Goldene, graphene’s golden cousin produced for the first time
-
 Stronger evidence links sedentary behavior and frailty in old age
Stronger evidence links sedentary behavior and frailty in old age
-
 Electronic socks prevent foot-related complications in diabetic patients
Electronic socks prevent foot-related complications in diabetic patients
Features

How the bias in algorithms can help us spot our own
People recognize their own biases in algorithms’ decisions more than they do in their own—even when those decisions are the same.
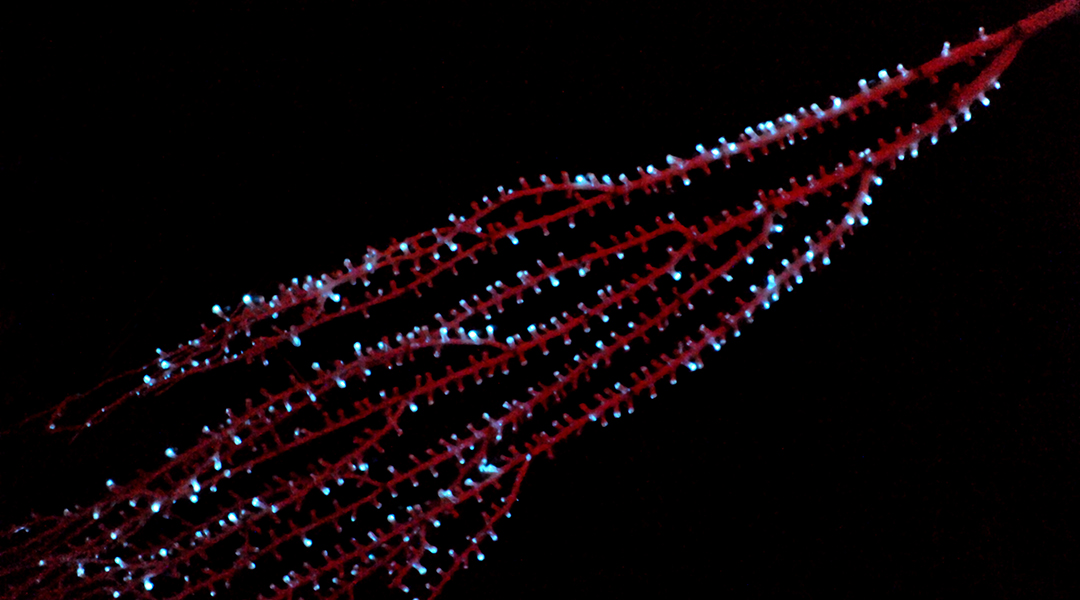
Bioluminescence evolved 300 million years earlier than previously thought
Bioluminescence has evolved independently in species time and again, but why this happened and when it first appeared has been a mystery.

Stronger evidence links sedentary behavior and frailty in old age
Data gathered from hundreds of thousands of individuals finds strong link between sedentary behavior and becoming frail, simple changes can help.

Fluorine helps make PET plastic waste easier to recycle
Pre-activation of plastics with fluorine-containing molecules disrupts their stability, making them easier to break down and upcycle.

Could AI be the reason we haven’t encountered alien civilizations?
A sensational paper argues that AI could be responsible for the scarcity of advanced technological civilizations in the Universe.
How the bias in algorithms can help us spot our own
People recognize their own biases in algorithms’ decisions more than they do in their own—even when those decisions are the same.
Bioluminescence evolved 300 million years earlier than previously thought
Bioluminescence has evolved independently in species time and again, but why this happened and when it first appeared has been a mystery.
Stronger evidence links sedentary behavior and frailty in old age
Data gathered from hundreds of thousands of individuals finds strong link between sedentary behavior and becoming frail, simple changes can help.
Most read this week
-
Quantum dot laser set to transform medicine and communications
-
Turning industrial waste into clean hydrogen fuel
-
Goldene, graphene’s golden cousin produced for the first time
-
Stronger evidence links sedentary behavior and frailty in old age
-
Electronic socks prevent foot-related complications in diabetic patients
ASN Weekly
Sign up for our weekly newsletter and receive the latest science news directly to your inbox.
Research news
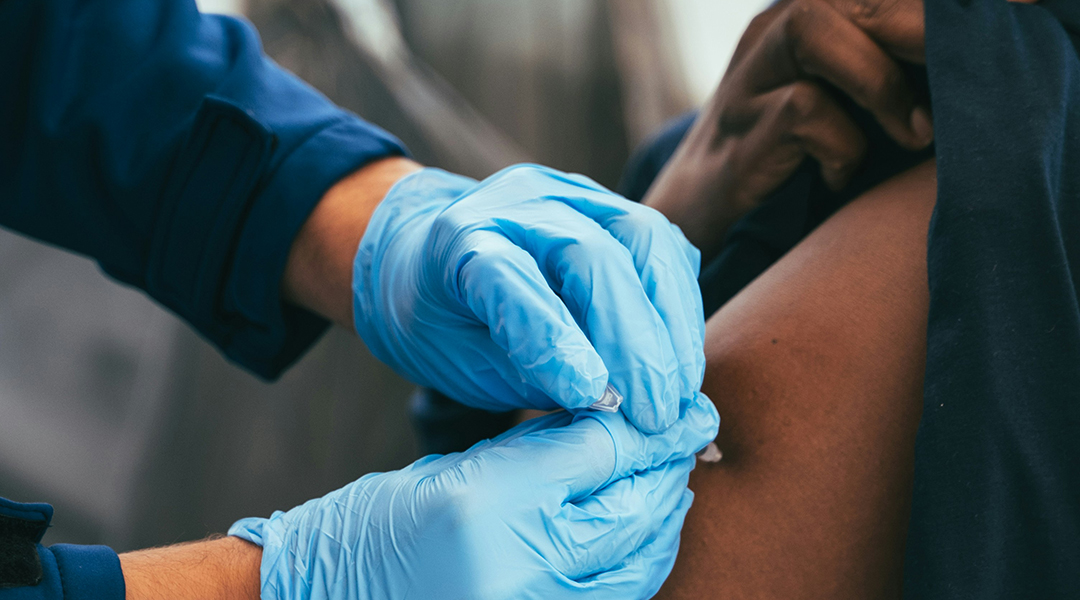
A universal vaccine could eliminate the need to track evolving strains
An RNA-based vaccine approach that is effective against all virus strains and safe for infants and immunocompromised individuals.

Pulling clean hydrogen fuel from seawater
A new electrolysis device could transform the way we produce hydrogen fuel from seawater, addressing challenges that hindered this process.

Quantum-proofing passwords and artwork with DNA encryption
Chaotic pools of DNA could be the future of encryption, proving authenticity of artwork or securing passwords against quantum computers.

Quantum dot laser set to transform medicine and communications
Quantum dots are key to a new laser that could transform medical imaging, diagnostics, and boost communication.

Electronic socks prevent foot-related complications in diabetic patients
An electronic sock detects an “unhealthy” walking style linked with diabetes and poor circulation to prevent foot ulcers and amputation.

Turning industrial waste into clean hydrogen fuel
New technique uses waste metal shavings to catalyze hydrogen production, turning nothing but trash and water into clean, renewable fuel.

Goldene, graphene’s golden cousin produced for the first time
Scientists have managed to create sheets of gold only a single atom thick using a hundred-year-old Japanese smithing method.
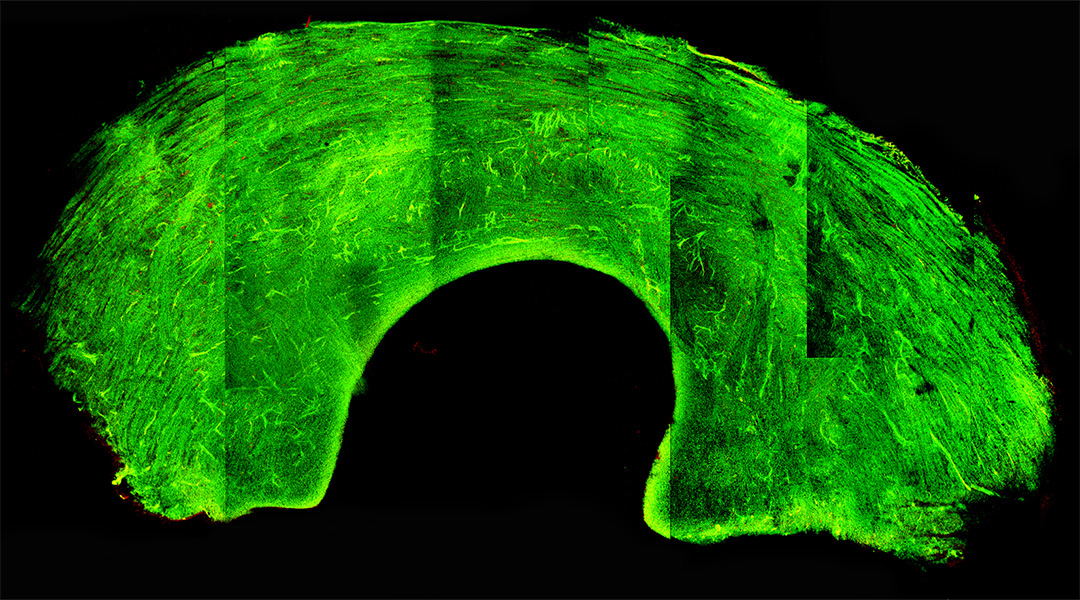
Glass-like knee grafts help address donor shortage
A cryopreservation technique puts graft tissue into a glass-like state, preserving cells and viability during long-term storage.
Fluorine helps make PET plastic waste easier to recycle
Pre-activation of plastics with fluorine-containing molecules disrupts their stability, making them easier to break down and upcycle.

Laser light induces magnetism at room temperature
Scientists create magnetism in a non-magnet at room temperature for the first time, with implications in quantum tech and computer science.
Could AI be the reason we haven’t encountered alien civilizations?
A sensational paper argues that AI could be responsible for the scarcity of advanced technological civilizations in the Universe.
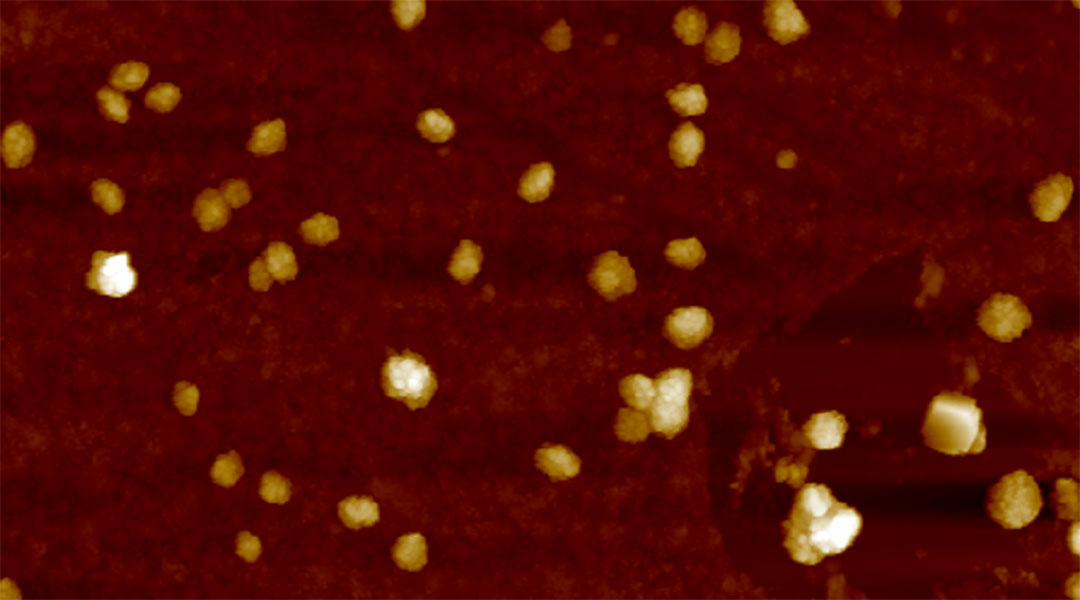
Peptides light up in the brain for early diagnosis of Alzheimer’s
Peptide-laden vesicles light up in the presence of amyloid beta, providing an early diagnostic test (and possible treatment) for Alzheimer’s.
Mushrooms could be the next big thing in energy storage
Scientists are using carbon filaments from mushrooms in supercapacitors, paving the way for a sustainable energy future.
Research news
A universal vaccine could eliminate the need to track evolving strains
An RNA-based vaccine approach that is effective against all virus strains and safe for infants and immunocompromised individuals.
Pulling clean hydrogen fuel from seawater
A new electrolysis device could transform the way we produce hydrogen fuel from seawater, addressing challenges that hindered this process.
Quantum-proofing passwords and artwork with DNA encryption
Chaotic pools of DNA could be the future of encryption, proving authenticity of artwork or securing passwords against quantum computers.
Quantum dot laser set to transform medicine and communications
Quantum dots are key to a new laser that could transform medical imaging, diagnostics, and boost communication.
Electronic socks prevent foot-related complications in diabetic patients
An electronic sock detects an “unhealthy” walking style linked with diabetes and poor circulation to prevent foot ulcers and amputation.
Turning industrial waste into clean hydrogen fuel
New technique uses waste metal shavings to catalyze hydrogen production, turning nothing but trash and water into clean, renewable fuel.
Goldene, graphene’s golden cousin produced for the first time
Scientists have managed to create sheets of gold only a single atom thick using a hundred-year-old Japanese smithing method.
Glass-like knee grafts help address donor shortage
A cryopreservation technique puts graft tissue into a glass-like state, preserving cells and viability during long-term storage.
Fluorine helps make PET plastic waste easier to recycle
Pre-activation of plastics with fluorine-containing molecules disrupts their stability, making them easier to break down and upcycle.
Laser light induces magnetism at room temperature
Scientists create magnetism in a non-magnet at room temperature for the first time, with implications in quantum tech and computer science.
Could AI be the reason we haven’t encountered alien civilizations?
A sensational paper argues that AI could be responsible for the scarcity of advanced technological civilizations in the Universe.
Peptides light up in the brain for early diagnosis of Alzheimer’s
Peptide-laden vesicles light up in the presence of amyloid beta, providing an early diagnostic test (and possible treatment) for Alzheimer’s.
Mushrooms could be the next big thing in energy storage
Scientists are using carbon filaments from mushrooms in supercapacitors, paving the way for a sustainable energy future.
How a gut-on-a-chip is getting to the bottom of our gut’s microbiome
This artificial gut will allow scientists to gain deeper insights into the biome that exists there and how dysregulation can lead to disease.

Athina Anastasaki: New ways to recycle old polymers
Polymer chemist Athina Anastasaki talks about establishing her career, inroads into polymer recycling, and resilience in academia.

How research into existential risk will help safeguard humanity
Florian Jehn combines pragmatism with optimism when considering potential threats to human civilization.

Michael Dickey, our guide through the surprising world of liquid metals
The chemical and biomolecular engineer delves into the versatile applications, surprising properties, and future possibilities of liquid metals.
How a gut-on-a-chip is getting to the bottom of our gut’s microbiome
This artificial gut will allow scientists to gain deeper insights into the biome that exists there and how dysregulation can lead to disease.
Athina Anastasaki: New ways to recycle old polymers
Polymer chemist Athina Anastasaki talks about establishing her career, inroads into polymer recycling, and resilience in academia.
How research into existential risk will help safeguard humanity
Florian Jehn combines pragmatism with optimism when considering potential threats to human civilization.